Fehlfunktionen hemmender Synapsen als Ursache neurologischer Erkrankungen
Forschungsbericht (importiert) 2008 - Max-Planck-Institut für Hirnforschung
Autoren
Eulenburg, Volker; Betz, Heinrich
Abteilungen
Neurochemie (Prof. Dr. Heinrich Betz) MPI für Hirnforschung, Frankfurt/Main
Zusammenfassung
Glyzin und GABA sind die beiden wichtigsten hemmenden Botenstoffe im zentralen Nervensystem. Störungen der hemmenden Erregungsübertragung konnten bereits mit neurologischen Erkrankungen wie Epilepsie oder Hyperekplexie assoziiert werden. Durch die Analyse gentechnisch veränderter Mäuse wurden nun zwei neue Genorte, das Collybistin- und das Glyzintransporter 2-Gen, als an diesen Krankheiten beteiligt identifiziert. Genetische Untersuchungen an Patienten belegen, dass beide Genorte in der Tat Krankheitsgene beim Menschen darstellen.
Einleitung
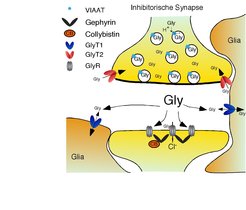
Die Aminosäure Glyzin ist neben der γ-Aminobuttersäure (GABA) im zentralen Nervensystem der wichtigste hemmende Botenstoff (Neurotransmitter). Glyzin und GABA werden in den Nervenendigungen in kleinen Membranbläschen, den sog. synaptischen Vesikeln, gespeichert (Abb. 1). Bei Erregung der Nervenzelle verschmelzen diese Vesikel mit der Zellmembran und schütten so ihren Inhalt auf die Zielneurone aus. Hier binden beide Neurotransmitter an spezifische Rezeptoren und bewirken so eine Hemmung (Inhibition) dieser Zellen. Diese wird durch die Wiederaufnahme der Neurotransmitter mittels hochaffiner Transportproteine in die Nervenendigung oder in umgebende Stützzellen (Glia) beendet.
Abb. 1: Schematische Darstellung einer inhibitorischen Synapse. An inhibitorischen Synapsen wird Glyzin in der vorgeschalteten Nervenendigung in Membranvesikeln gespeichert. Nach Erregung verschmelzen diese in einem Calcium-abhängigen Prozess mit der Zellmembran und schütten ihren Inhalt auf die Zielzelle aus. Das so freigesetzte Glyzin bindet an der nachgeschalteten Zelle an spezifische Rezeptoren, die durch das Protein Gephyrin an der Synapse verankert sind. Dies aktiviert und öffnet den Rezeptorionenkanal, durch den Chlorid in die Zelle einströmt kann und so eine Hemmung der Zielzelle bewirkt. Anschließend wird das Glyzin durch hochaffine Transporter (GlyT1 und GlyT2) aus der Synapse entfernt und die Hemmung des Zielneurons beendet.
Abb. 1: Schematische Darstellung einer inhibitorischen Synapse. An inhibitorischen Synapsen wird Glyzin in der vorgeschalteten Nervenendigung in Membranvesikeln gespeichert. Nach Erregung verschmelzen diese in einem Calcium-abhängigen Prozess mit der Zellmembran und schütten ihren Inhalt auf die Zielzelle aus. Das so freigesetzte Glyzin bindet an der nachgeschalteten Zelle an spezifische Rezeptoren, die durch das Protein Gephyrin an der Synapse verankert sind. Dies aktiviert und öffnet den Rezeptorionenkanal, durch den Chlorid in die Zelle einströmt kann und so eine Hemmung der Zielzelle bewirkt. Anschließend wird das Glyzin durch hochaffine Transporter (GlyT1 und GlyT2) aus der Synapse entfernt und die Hemmung des Zielneurons beendet.
Aus früheren Studien an Mäusen ist bekannt, dass Störungen der Erregungsübertragung an inhibitorischen Synapsen schwerwiegende Konsequenzen für die Funktion des Nervensystems haben (Übersicht in [1]). So führen zum Beispiel Mutationen in den Rezeptorproteinen, die die Hemmung von Zielzellen durch GABA oder Glyzin vermitteln, zu Epilepsie beziehungsweise Hyperekplexie, das heißt zu Erkrankungen, welche auf Übererregbarkeit von Nervenzellen im Gehirn bzw. Rückenmark zurückzuführen sind [1; 2]. Auch die Inaktivierung von Genen für Ankerproteine, welche die Rezeptoren an der Synapse halten, oder für Transporter, die die Neurotransmitter nach erfolgter Signalweiterleitung in die umliegenden Zellen schaffen, verursachen schwere Fehlfunktionen der jeweiligen Synapsen. Zum Beispiel zeigen Mäuse, denen das Protein Gephyrin fehlt, welches für die synaptische Verankerung von GABA- und Glyzin-Rezeptoren verantwortlich ist, erhebliche Veränderungen in der Zahl und im Erregungszustand der die Muskulatur innervierenden Nervenzellen im Hirnstamm und Rückenmark – die Tiere sterben am ersten Tag nach der Geburt aufgrund von Schluck- und Atemstörungen [3; 4]. Vergleichbare Symptome zeigen Mäuse, die Mutationen in Glyzinrezeptor-Genen tragen [5; 6] oder denen der Glyzintransporter 2 (GlyT2) fehlt [7].
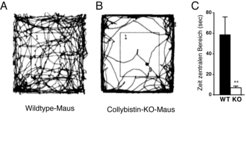
Abb. 2: Angstverhalten von Collybistin-defizienten (KO) Mäusen. Mäuse, denen die kleine GTPase Collybistin fehlt, und deren Wildtyp-Geschwister wurden in einen neuen, den Mäusen unbekannten Käfig gesetzt und ihr Bewegungsverhalten während der nächsten 10 min registriert. Während Wildtyp-Mäuse das komplette ihnen zur Verfügung stehende Areal explorieren, bleiben Collybistin-KO-Mäuse in den Randbereichen des Käfigs und besuchen den weniger geschützten mittleren Bereich des Käfigs nur sporadisch. Dieses Verhalten zeigt, dass Collybistin-KO-Mäuse ängstlicher sind als ihre genetisch unveränderten Geschwister.
Abb. 2: Angstverhalten von Collybistin-defizienten (KO) Mäusen. Mäuse, denen die kleine GTPase Collybistin fehlt, und deren Wildtyp-Geschwister wurden in einen neuen, den Mäusen unbekannten Käfig gesetzt und ihr Bewegungsverhalten während der nächsten 10 min registriert. Während Wildtyp-Mäuse das komplette ihnen zur Verfügung stehende Areal explorieren, bleiben Collybistin-KO-Mäuse in den Randbereichen des Käfigs und besuchen den weniger geschützten mittleren Bereich des Käfigs nur sporadisch. Dieses Verhalten zeigt, dass Collybistin-KO-Mäuse ängstlicher sind als ihre genetisch unveränderten Geschwister.
In Zusammenarbeit mit Arbeitsgruppen in Braunschweig und Göttingen konnten Wissenschaftler am Max-Planck-Institut für Hirnforschung mit Collybistin ein weiteres Protein identifizieren, das für die Funktion von inhibitorischen Synapsen wichtig ist. Collybistin gehört zu einer großen Familie intrazellulärer Signalproteine, den so genannten kleinen GTPasen, die viele zelluläre Prozesse regulieren [8]. Durch die Herstellung von sog. Knockout-Mäusen, in denen das Collybistin-Gen inaktiviert ist, konnten die Forscher zeigen, dass Collybistin für die synaptische Lokalisation von Gephyrin und bestimmten inhibitorischen GABAA-Rezeptorsubtypen in Vorderhirnbereichen wie dem Hippocampus und den Mandelkernen (Amygdala), nicht aber im Rückenmark, wichtig ist [9; 10]. Dies zeigt, dass inhibitorische Rezeptoren über unterschiedliche Regulationsmechanismen an Synapsen verankert werden können. Funktionell wird die Collybistin-Defizienz bei Mäusen an einer Reduktion der hemmenden Erregungsübertragung sichtbar [9]. Auf der Verhaltensebene bewirkt sie neben einer milden Form von Epilepsie auch Störungen im Angst- und Lernverhalten [9], wie Abbildung 2 zeigt.
Dass diese in Mausmodellen erhaltenen Befunde auch für humane Erkrankungen relevant sein können, konnte in der Vergangenheit für viele Mutationen in Glyzinrezeptor-Genen gezeigt werden (Übersicht in [2]). Hier existiert ein direkter Zusammenhang zwischen dem Auftreten der vererbbaren Form der sog. Hyperekplexie („Schreckkrankheit“) und bestimmten Mutationen in den Rezeptorgenen. Aber auch für die anderen oben genannten Gene des inhibitorischen Synapsenapparates wurde inzwischen nachgewiesen, dass sie Krankheitsgene beim Menschen sein können. So erschienen im letzten Jahr zwei humangenetische Arbeiten, in denen berichtet wird, dass Mutationen bzw. Defekte im Collybistin-Gen Formen des Schwachsinns zugrunde liegen, die ähnlich wie bei den Collybistin-defizienten Mäusen u.a. mit erhöhter Ängstlichkeit, veränderter Motorik und Anfällen assoziiert sind [10; 11].
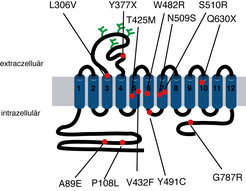
Abb. 3: Membrantopologie des humanen GlyT2. Positionen, in denen Mutationen bei Hyperekplexie-Patienten gefunden wurden, sind in der Figur angegeben (rote Punkte). Die Art der Substitution ist zusätzlich angegeben.
Abb. 3: Membrantopologie des humanen GlyT2. Positionen, in denen Mutationen bei Hyperekplexie-Patienten gefunden wurden, sind in der Figur angegeben (rote Punkte). Die Art der Substitution ist zusätzlich angegeben.
Da GlyT2-defiziente Mäuse eine ähnliche Symptomatik wie Hyperekplexie-Patienten (verstärkte Schreckreaktion, spontaner Tremor sowie spastische Muskelkrämpfe) aufweisen, haben die Wissenschaftler um Eulenburg und Betz untersucht, ob Mutationen im GlyT2-Gen auch für die Entstehung humaner Formen der Hyperekplexie verantwortlich sein könnten. In der Tat konnten in mehreren Hyperekplexie-Patienten, bei denen keine Mutationen in Glyzinrezeptor-Genen gefunden wurden, Mutationen im GlyT2-Gen identifiziert werden, die zumindest teilweise zu defekten Transporterproteinen führen [13]. Diese Assoziation von GlyT2-Mutationen mit der humanen Erbkrankheit Hyperekplexie wurde von einer weiteren Arbeitsgruppe bestätigt und erweitert [14], sodass Mutationen im GlyT2-Gen neben Mutationen in den Glyzinrezeptor-Genen heute zu den häufigsten genetischen Ursachen für Hyperekplexie gezählt werden (Abb. 3).
Zusammenfassend zeigen all diese Untersuchungen, dass Defekte der inhibitorischen Neurotransmission Ursache schwerer Erkrankungen bei Tier und Mensch sein können. Die Wissenschaftler erwarten, dass die Identifizierung neuer, an der hemmenden Erregungsübertragung beteiligter Gene und Proteine weitere humanrelevante Pathogenesemechanismen aufdecken wird. Deren molekulare Untersuchung ist wichtig, weil sich darüber auch neue Therapieansätze erschließen lassen. Für eine Glyzinrezeptormutation, die beim Menschen Hyperekplexie verursacht, konnten die Forscher zeigen, dass die Regelfunktion des mutierten Rezeptors in vitro und in einem Mausmodell zumindest vorübergehend durch klinisch vielfach eingesetzte Anästhetika „geheilt“ werden kann [15].
Originalveröffentlichungen
1.
H. Möhler:
GABAA receptors in central nervous system disease: anxiety, epilepsy, and insomnia.
Journal of Receptors and Signal Transduction 26, 731-740 (2006).
2.
R. J. Harvey, M. Topf, K. Harvey, M. I. Rees:
The genetics of hyperekplexia: more than startle!
Trends in Genetics 24, 439-47 (2008).
3.
G. Feng, H. Tintrup, J. Kirsch, M. C. Nichol, J. Kuhse, H. Betz, J. R. Sanes:
Dual requirement for gephyrin in glyine receptor clustering and molybdoenzyme activity.
Science 282, 1321-1324 (1998).
4.
G. B. Banks, R. Kanjhan, S. Wiese, M. Kneussel, L. M. Wong, G. O’Sullivan, M. Sendtner, M. C. Bellingham, H. Betz, P. G. Noakes:
Glycinergic and GABAergic synaptic activity differentially regulate motoneuron survival and skeletal muscle innervation.
Journal of Neuroscience 25, 1249-1259 (2005).
5.
C.-M. Becker, V. Schmieden, P. Tarroni, U. Strasser, H. Betz:
Isoform-selective deficit of glycine receptors in the mouse mutant spastic.
Neuron 8, 283-289 (1992).
6.
C. Mülhardt, M. Fischer, P. Gass, D. Simon-Chazottes, J.-L. Guénet, J. Kuhse, H. Betz, C-M. Becker:
The spastic mouse: aberrant splicing of glycine receptor β subunit mRNA caused by intronic insertion of L1 element.
Neuron 13, 1003-1015 (1994).
7.
J. Gomeza, K. Ohno, S. Hülsmann, W. Armsen, V. Eulenburg, D. W. Richter, B. Laube, H. Betz:
Deletion of the mouse glycine transporter 2 results in a hyperekplexia phenotype and postnatal lethality.
Neuron 40, 797-806 (2003).
8.
S. Kins, H. Betz, J. Kirsch:
Collybistin, a novel brain-specific GEF, induces submembrane clustering of gephyrin.
Nature Neuroscience 3, 22-29 (2000).
9.
T. Papadopoulos, M. Korte, V. Eulenburg, H. Kubota, M. Retiounskaia, R. J. Harvey, K. Harvey, G. A. O’Sullivan, B. Laube, S. Hülsmann, J. R. P. Geiger, H. Betz:
Impaired GABAA receptor clustering and altered hippocampal synaptic plasticity in collybistin-deficient mice.
EMBO Journal 26, 3888-3899 (2007).
10.
T. Papadopoulos, V. Eulenburg, S. Reddy Alla, I. M. Mansuy, Y. Li, H. Betz:
Collybistin is required for both the formation and maintenance of GABAergic postsynapses in the hippocampus.
Molecular and Cellular Neuroscience 39, 161-169 (2008).
11.
E. J. Marco, F. E. Abidi, J. Bristow, W. B. Dean, P. Cotter, R. J. Jeremy, C. E. Schwartz, E. H. Sherr:
ARHGEF9 disruption in a female patient is associated with X-linked mental retardation and sensory hyperarousal.
Journal of Medical Genetics 45, 100-105 (2008).
12.
V. M. Kalscheuer, L. Musante, C. Fang, K. Hoffmann, C. Fuchs , E. Carta, E. Deas, K. Venkateswarlu, C. Menzel, R. Ullmann, N. Tommerup, L. Dalprà, A. Tzschach, A. Selicorni, B. Lüscher, H.H. Ropers, K. Harvey, R.J. Harvey:
A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression, and mental retardation.
Human Mutation 30, 61-8 (2009).
13.
V. Eulenburg, K. Becker, J. Gomeza, B. Schmitt, C.-M. Becker, H. Betz:
Mutations within the human GlyT2 (SLC6A5) gene associated with hyperekplexia.
Biochemical and Biophysical Research Communications 348, 400-405 (2006).
14.
M. I. Rees, K. Harvey, B. R. Pearce, S. K. Chung, I. C. Duguid, P. Thomas, S. Beatty, G. E. Graham, L. Armstrong, R. Shiang, K. J. Abbott, S. M. Zuberi, J.B. Stephenson, M.J. Owen, M.A. Tijssen, A.M. v.d. Maagdenberg, T.G. Smart, S. Supplisson, R.J. Harvey:
Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of
human startle disease.
Nature Genetics 38, 801-806 (2006).
15.
S. M. O'Shea, L. Becker, H. Weiher, H. Betz, B. Laube:
Propofol restores the function of "hyperekplexic" mutant glycine receptors in Xenopus oocytes and mice.
 aus der Synapse entfernt und die Hemmung des Zielneurons beendet.")
 Mäusen. Mäuse, denen die kleine GTPase Collybistin fehlt, und deren Wildtyp-Geschwister wurden in einen neuen, den Mäusen unbekannten Käfig gesetzt und ihr Bewegungsverhalten während der nächsten 10 min registriert. Während Wildtyp-Mäuse das komplette ihnen zur Verfügung stehende Areal explorieren, bleiben Collybistin-KO-Mäuse in den Randbereichen des Käfigs und besuchen den weniger geschützten mittleren Bereich des Käfigs nur sporadisch. Dieses Verhalten zeigt, dass Collybistin-KO-Mäuse ängstlicher sind als ihre genetisch unveränderten Geschwister.")
. Die Art der Substitution ist zusätzlich angegeben.")