Molekulare Analyse der synaptischen Hemmung
Forschungsbericht (importiert) 2003 - Max-Planck-Institut für Hirnforschung
Die Aminosäure Glyzin ist neben der γ-Aminobuttersäure (GABA) die wichtigste hemmende Überträgersubstanz (Neurotransmitter) im zentralen Nervensystem. Glyzin wird in Nervenendigungen in kleinen sog. synaptischen Vesikeln gespeichert und bei Erregung an den Schaltstellen zwischen Nervenzellen, den Synapsen, auf Zielneurone ausgeschüttet. Dort bindet es an spezifische Glyzinrezeptoren, welche porenartige Ionenkanäle in der Zellmembran von Neuronen ausbilden. Nach Bindung des Transmitters Glyzin öffnet sich der Rezeptor-Kanal, und Chlorid-Ionen strömen in die Zelle ein. Dieser Ionenfluss hemmt die Nervenreizleitung und dämpft das "Feuern" der Neurone. Nach der Rezeptorbindung wird Glyzin durch hochaffine Transportsysteme, die sog. Glyzintransporter, in die Nervenendigung bzw. umliegenden Gliazellen aufgenommen und so der Übertragungsvorgang beendet.
Für Glyzin sind im Zentralnervensystem von Säugetieren zwei verschiedene Transporterproteine bekannt. Das eine (GlyT1) ist in den die Nervenzellen umgebenden Stütz- oder Gliazellen lokalisiert, das zweite (GlyT2) hochspezifisch in Hemmung vermittelnden Interneuronen des Rückenmarks und Hirnstamms exprimiert. Um die spezifischen Funktionen dieser Glyzintransporter präziser zu erhellen, wurden Mauslinien entwickelt, in denen die GlyT1- bzw. GlyT2-Gene inaktiviert sind. Diese Mauslinien stellen Tiermodelle für zwei seltene neurologische Erbkrankheiten des Menschen dar, die so genannte Glyzinenzephalopathie, welche zu einer schweren Störung der Hirnentwicklung und Tod im frühen Kindesalter führt, und die hereditäre Hyperekplexie oder "Schreckkrankheit", deren schwere Formen ebenfalls mit frühem Kindstod einhergehen können. Die Untersuchung der Mutantentiere ergab, dass GlyT1 hauptsächlich für die Terminierung der glyzinergen Neurotransmission verantwortlich ist, während GlyT2 die Wiederaufnahme von Glyzin in die vorgeschaltete Nervenendigung vermittelt und dadurch die Bereitstellung von Glyzin für neue Übertragungsvorgänge sichert. Somit haben gliale und neuronale Transporter unterschiedliche Funktionen.
Die an den Glyzintransporter-defizienten Mauslinien gewonnenen Erkenntnisse sind für die Entwicklung neuer therapeutischer Ansätze von Bedeutung. Das GlyT1-Protein z. B. gilt derzeit als eines der wichtigsten Zielmoleküle für die Entwicklung neuer bei Schizophrenie wirksamer Pharmaka. Die genauen Ursachen dieser häufigsten psychiatrischen Erkrankung sind bisher ungeklärt. In Tierversuchen und klinischen Studien an Schizophreniepatienten konnte aber gezeigt werden, dass die verbesserte Aktivierung des Glyzin-abhängigen erregenden NMDA-Rezeptors durch direkte Glyzinsubstitution oder pharmakologische Hemmung der GlyT1-vermittelten Glyzinaufnahme typische Symptome der Schizophrenie deutlich reduziert. Etliche Forschungslabors in Pharmafirmen suchen deshalb nach klinisch nutzbaren Inhibitoren von GlyT1, da solche Stoffe für die Behandlung von Psychosen vielversprechend erscheinen. Die jetzt durch Geninaktivierung erhaltenen Befunde lassen vermuten, dass derartige Therapieansätze möglicherweise mit erheblichen Nebenwirkungen behaftet sind, da GlyT1 vitale Funktionen an hemmenden Synapsen im Hirnstamm hat. In neugeborenen GlyT1-defizienten Tieren führt die verminderte Glyzinaufnahme zu einer verstärkten Aktivierung hemmender Rezeptoren, wodurch es zu einer lethalen Suppression des Atemrhythmus kommt.
In Zusammenarbeit mit der Arbeitsgruppe Müller, der Abteilung Neuroanatomie und vielen Kollegen aus dem In- und Ausland gelang außerdem bei Arbeiten an einem Subtyp des inhibitorischen Glyzinrezeptores, GlyRα3, die Identifikation eines bisher nicht bekannten Steuermechanismus bei der Schmerzverarbeitung. Mithilfe genetisch veränderter Mäuse konnte jener molekulare Signalweg aufgeklärt werden, der bei Entzündungen zu einer verstärkten Schmerzreizleitung vom Rückenmark zum Gehirn führt. Entzündungsreaktionen, die als Folge einer Gewebeverletzung entstehen können, gehen typischerweise mit einer erhöhten Empfindlichkeit gegenüber Schmerzen einher. Hierbei führen Reize, die bereits unter Normalbedingungen als schmerzhaft empfunden werden, zu einer massiv verstärkten Schmerzreaktion (Hyperalgesie), und an sich neutrale Reize, wie beispielsweise leichte Berührungen, können heftige Schmerzen auslösen (Allodynie). Diese inflammatorische Sensitisierung gegenüber Schmerzreizen ist z. T. auf eine erhöhte Erregbarkeit der peripheren Nervenzellen zurückzuführen. Forschungsarbeiten der letzten Jahre zeigen aber, dass auch Nervenzellen des Rückenmarks durch entzündliche Prozesse empfindlicher werden.
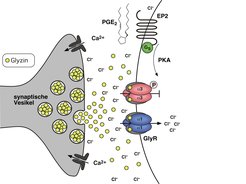
Hierbei spielen Prostaglandine, eine Klasse von Botenstoffen, die während Entzündungsreaktionen ausgeschüttet werden, eine zentrale Rolle. So beruht die Wirkungsweise von Schmerzmitteln wie Aspirin auf der Hemmung der Prostaglandin-Synthese. Interessanterweise werden Prostaglandine, insbesondere der Subtyp E2 (PGE2), nicht nur von Zellen in der unmittelbaren Umgebung des Entzündungsherdes produziert, sondern auch im Rückenmark steigt der Prostaglandin-Spiegel massiv an. Das eingehende Schmerzsignal wird somit im Rückenmark über eine PGE2-abhängige Signalkaskade nochmals verstärkt. Diese Verstärkung erfolgt in den oberflächlichen Schichten des Rückenmarks, dem dorsalen Horn. Dieses ist die zentrale Schaltstelle zwischen schmerzempfindlichen Nervenfasern der Peripherie und nachgeschalteten Nervenzellen, die das Signal dann zum Gehirn weiterleiten.
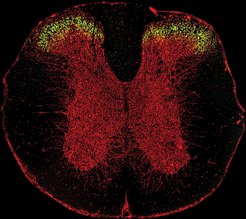
Ziel der Arbeit war ursprünglich, die Funktion des GlyRα3-Subtyps zu verstehen. Hierzu wurden Gewebeschnitte des Rückenmarks von Mäusen mit Fluoreszenz-markierten Antikörpern gegen GlyRα3 inhibiert (vgl. Abb. 1). Diese Versuche ergaben für GlyRα3 eine spezifische Lokalisation im dorsalen Horn des Rückenmarks, was auf eine mögliche Funktion des Rezeptors in der Schmerzreizleitung hinwies. Aus elektrophysiologischen Untersuchungen der Arbeitsgruppe von Hanns-Ulrich Zeilhofer von der Universität Erlangen-Nürnberg war bekannt, dass die Nervenimpulsleitung von Neuronen des dorsalen Horns durch Prostaglandine moduliert wird und dass hierbei Glyzin-Rezeptoren beteiligt sind. Mithilfe von genetisch modifizierten Mäusen, in denen das Gen für GlyRα3 inaktiviert wurde, wurden in Zusammenarbeit mit Zeilhofer gezeigt, dass spezifisch die α3-Isoform des Glyzinrezeptors von Prostaglandinen moduliert wird und diesen Rezeptoren damit eine Schlüsselfunktion in der inflammatorischen Schmerzsensitisierung zukommt.
 und Gephyrin (rot) zeigt die spezifische Lokalisation von GlyRα3 im dorsalen Horn.")
So zeigten elektrophysiologische Messungen an Rückenmarksschnitten von GlyRα3-defizienten Tieren nicht die typische Hemmung der Glyzin-abhängigen Nervenreizleitung. Um herauszufinden, welche Rolle GlyRα3 in der physiologischen Schmerzantwort hat, wurde die Schmerzempfindlichkeit von GlyRα3-defizienten mit der von normalen Mäusen verglichen. Beide Tiergruppen reagierten in gleicher Weise auf akute Schmerzreize. Wurde jedoch zunächst eine Entzündungsreaktion, zum Beispiel durch Injektion einer irritierenden Substanz in die Pfote induziert, so blieb die hierdurch in normalen Mäusen ausgelöste, langanhaltende Schmerzsensitisierung in den GlyRα3-defizienten Tieren aus.
Zusammenfassend zeigen diese Versuche, dass die Hemmung von GlyRα3 essentiell ist für die nach Entzündungen gesteigerte Schmerzreizleitung durch Rückenmarksneurone. Auf molekularem Niveau löst die Bindung des Prostaglandins PGE2 an Zelloberflächenrezeptoren die Aktivierung einer Signalkaskade aus, die zur Übertragung von Phosphatgruppen auf GlyRα3 führt (vgl. Abb. 2). Hierdurch wird der Rezeptor inaktiviert und kann seine hemmende Wirkung auf die Schmerzreizleitung nicht mehr ausüben. Letztlich führt dies zu einer gesteigerten Aktivität der Rückenmarksneurone, das Schmerzsignal wird somit stärker. Die Entwicklung von Substanzen, welche diesen Signalweg unterbinden, könnte zu neuen Therapien für chronische Schmerzzustände führen.